
In a study published in Nature
Methods, a research team led by Prof. Qu Kun from the University of Science and
Technology of China (USTC) of the Chinese Academy of Sciences has benchmarked
16 spatial and single-cell transcriptomics integration methods for transcript
distribution prediction and cell type deconvolution.
The spatial location of cells in tissues and organs plays a crucial role in performing its specific functions. In recent years, researchers have developed various spatial transcriptomics approaches to detect whole-transcriptome-level data in cells while maintaining accurate spatial orientation. However, existing spatial transcriptomics approaches have two deficiencies. The approaches based on next-generation sequencing could not detect single-cell deconvolution of the spot. In addition, the approaches based on in situ hybridization and fluorescence microscopy are limited in the total number of RNA transcripts obtained.
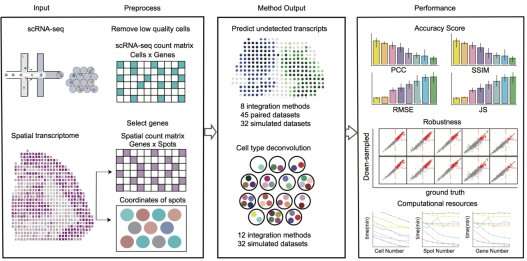
In view of this, researchers have developed various integration methods to combine spatial transcriptomic data with single-cell RNA-seq (scRNA-seq) data to predict the spatial distribution of undetected transcripts and/or perform cell type deconvolution of spots in histological sections. These integration methods have undoubtedly promoted understanding of spatial transcriptomics data and related biological and pathological processes. However, due to diversified operating principles and applicable scope, researchers had difficulty in selecting optimal integration methods.
The research team led by Prof. Qu has been working on developing analysis algorithms and software for biological big data for a long time. In this study, the researchers presented benchmarking of 16 integration methods using 45 paired datasets containing both spatial transcriptomics and scRNA-seq data and 32 simulated datasets according to their accuracy, robustness, and computational resources consumption.
According to the researchers, Cell2location, SpatialDWLS, and Robust Decomposition of Cell Type (RCTD) were superior to other integration methods for the cell type deconvolution of spots in histological sections, whereas Tangram, gimVI, and Spatial Gene Enhancement (SpaGE) showed excellent performance compared with other integration methods for predicting the spatial distribution of RNA transcripts. Tangram, Seurat and Linked Inference of Genomic Experimental Relationships (LIGER) had high computational efficiency and were suitable for processing large-scale data sets.
This study concludes the attribute, performance, and applicability of each integration method and the advantages of methods with high efficiency. It helps researchers to further improve these algorithms.
The team also shares a benchmark process of integrating spatial transcriptomics data with scRNA-seq data on GitHub to help researchers select optimal integration methods to process their datasets.
 Previous page
Previous page Back to top
Back to top


